Science
Pioneering Gene Therapy for Inherited Retinal Diseases
Inherited Retinal Dystrophies (IRDs) are a group of genetic disorders causing progressive vision loss. These conditions arise from mutations in genes crucial for photoreceptor and retinal pigment epithelium (RPE) cell function. Retinitis Pigmentosa, the most prevalent form, affects an estimated 1.5 million people globally.
Our research pushes the boundaries of treatment for rare and challenging outer retinal layer disorders. While traditional therapies focus on symptom management, we target the root cause. For Retinitis Pigmentosa, especially cases with Rhodopsin (RHO) gene mutations, we’re pioneering innovative approaches using genome editing and HITI (Homology-Independent Targeted Integration) gene insertion techniques.
In collaboration with Hiroshima University, we’ve optimized highly specific Zinc Finger Nucleases (ZFNs), achieving editing efficiencies comparable to CRISPR-Cas9. Our collaboration with Kobe Eye Center Hospital is advancing precise and efficient delivery methods for these therapeutic agents. These efforts are paving the way for more effective and durable treatments, offering new hope to those affected by debilitating conditions.
Inherited Retinal Dystrophy (IRD)
Inherited Retinal Dystrophy (IRD) is an umbrella term for a group of genetic eye disorders that cause the progressive degeneration of photoreceptor cells. Among these, Retinitis Pigmentosa is the most common, affecting an estimated 1.5 million people worldwide. Genetic testing conducted by leading institutions, including Kobe City Eye Center Hospital, identified approximately 100 cases associated with these conditions. Most of these genes play crucial roles in maintaining the health and function of the RPE and photoreceptor cells. When mutations occur in these genes, it can trigger a cascade of degeneration, ultimately leading to photoreceptor cell death.
Unfortunately, there is currently no cure for Retinitis Pigmentosa. Treatment strategies mainly focus on managing symptoms and slowing the disease progression. These approaches include prescribing medications to enhance dark adaptation and improve blood flow, recommending vitamin supplements, and providing specialized sunglasses to shield eyes from harsh light. Although these methods can help patients cope with their symptoms, they do not address the underlying root cause of the condition.
Structure of the Eye and Retina
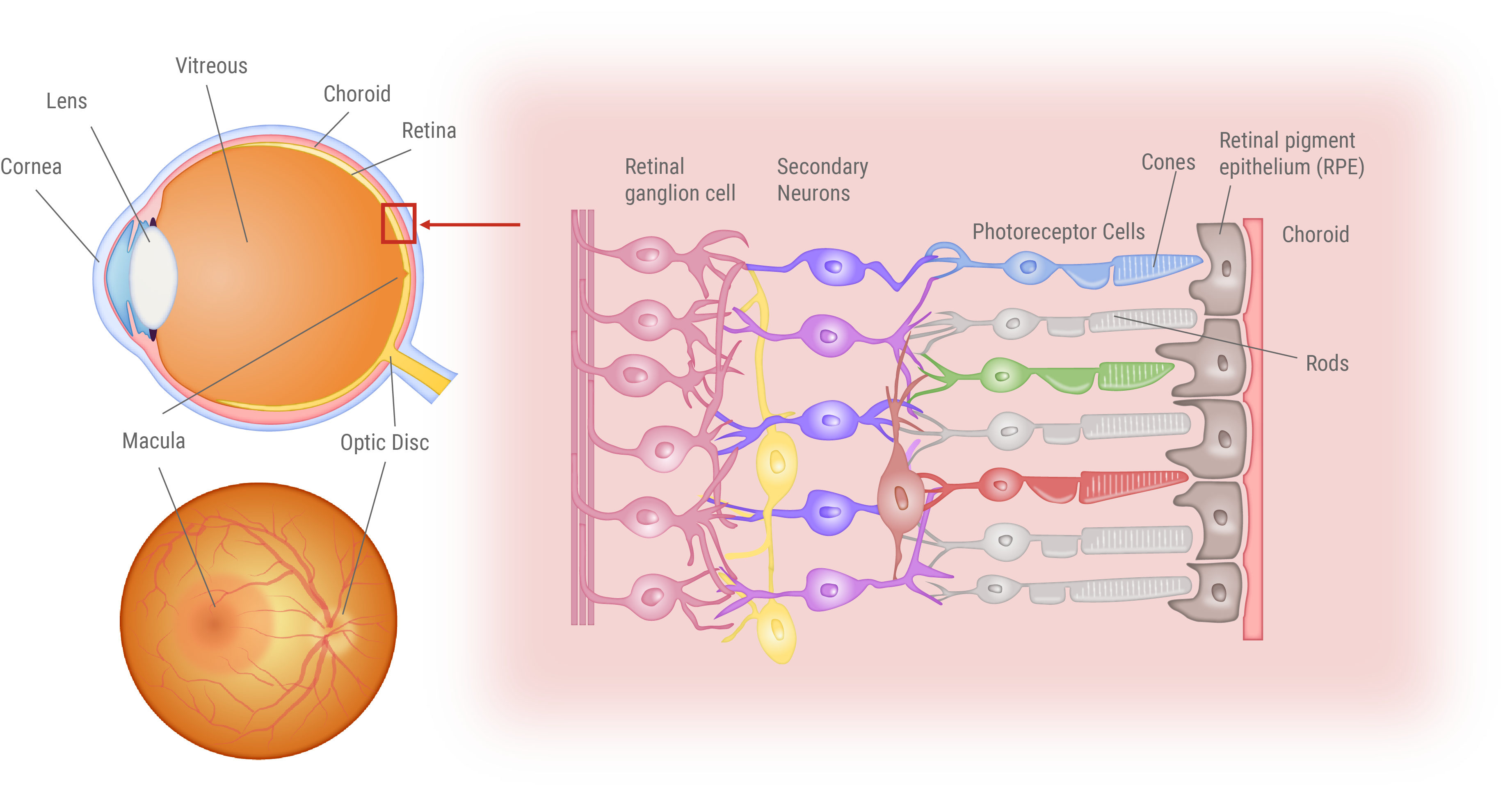
Visual Impairment in Retinitis Pigmentosa and Age-Related Macular Degeneration
This figure illustrates the distinct vision impairments experienced by patients with Retinitis Pigmentosa (RP) and Age-related Macular Degeneration (AMD). Patients endure a gradual loss of vision over time. For RP patients, the loss first occurs in the peripheral vision, progressively closing in on the center. In contrast, AMD patients first experience loss of central vision, making it challenging to focus on details.

Gene-Targeted Therapeutics
Recent advances in science and technology have revolutionized medical treatment, expanding beyond traditional small-molecule drugs to include a wide array of therapies using polymers, nucleic acids, and even cells. This diverse range of innovative therapies and drugs is collectively known as “drug/medical modalities.” Among these, gene therapy has emerged as a groundbreaking approach that harnesses the power of genes (such as DNA or RNA) or targets a patient’s own genome.
Gene therapy often refers to treatments that directly address the root cause of genetic diseases. With recent advancements in genetic diagnosis and medical infrastructure, there is a growing interest in the potential of these therapies, especially for genetic disorders with known causative genes. While conventional treatments such as small molecules or antibodies have limitations, gene therapies promise more comprehensive and targeted effects.
Our research focuses on developing cutting-edge treatments for the most common form of Retinitis Pigmentosa, particularly for Rhodopsin (RHO) gene-associated mutations. By targeting the genetic root of the disease, we aim to offer more effective solutions than ever before.
Targeted gene therapeutics
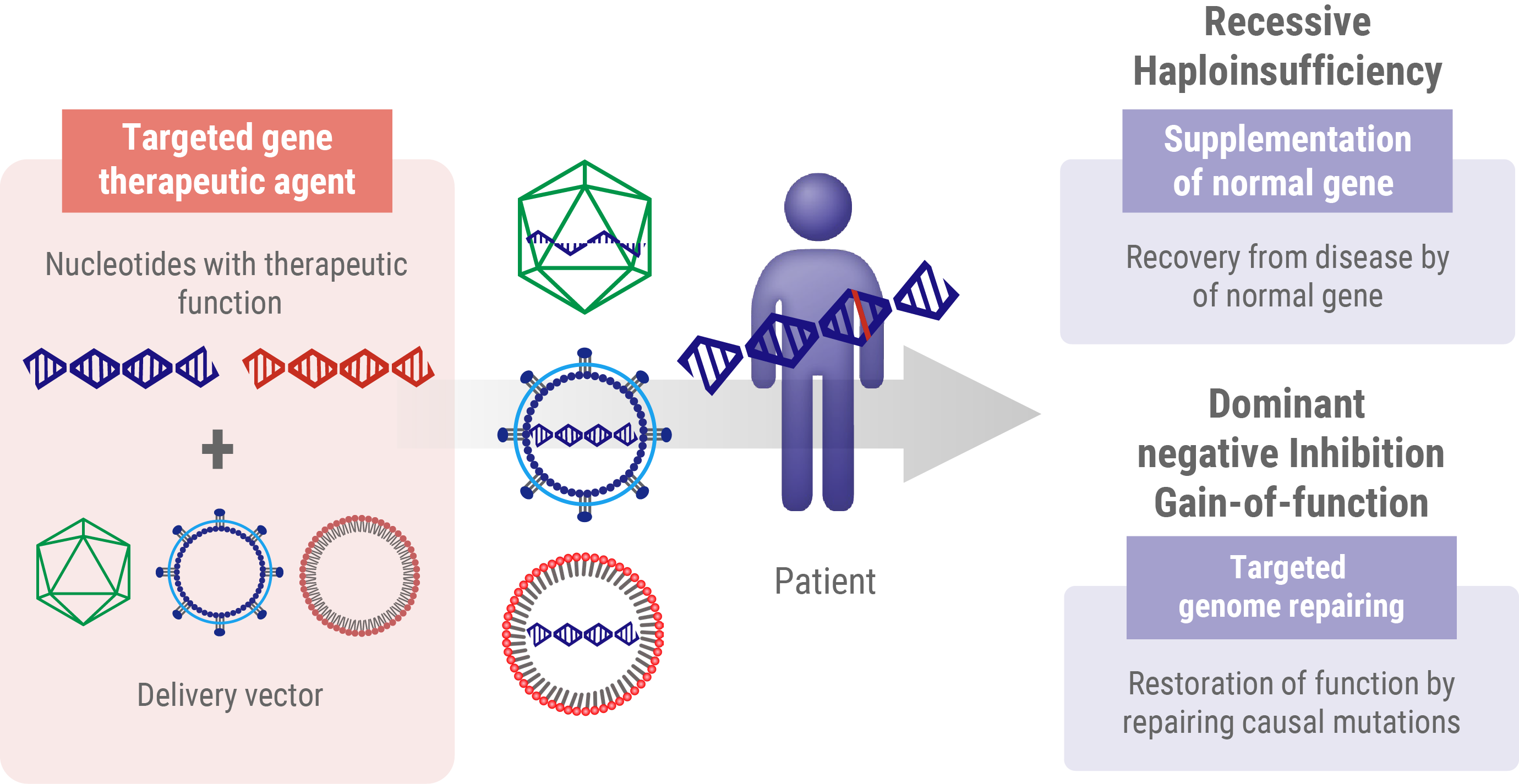
Gene Therapy for Recessive Mutations
In recessive mutations, the protein product does not work properly but does not interfere with normal protein function. Individuals with these recessive mutations only develop the disease when both alleles of a particular gene are mutated. In these cases, the go-to strategy in gene therapy is to introduce a working version of the gene from outside the body. This approach, known as gene supplementation, aims to fill the gap caused by loss-of-function gene products. Think of it like adding a fully functional spare part to a machine where both original parts are broken. By introducing this ‘spare part’ – the healthy gene – we’re aiming to restore the missing function and potentially slow or stop the progression of the disease.
Gene Therapy Strategies for Dominant Mutations
- Haploinsufficiency: In this scenario, a single functional allele is insufficient to maintain a normal physiological function. The therapeutic approach for haploinsufficiency is analogous to that used for recessive mutations, which involves the introduction of a wild-type gene to supplement inadequate protein production.
- Dominant-negative (or gain-of-function) mutations These mutations result in the production of an aberrant protein, which interferes with the function of the wild-type protein. Addressing dominant-negative mutations requires more sophisticated genome editing techniques to either correct or nullify the mutated allele.
Understanding these nuanced differences in mutation types and their effects is fundamental for developing effective, tailored gene therapy interventions for genetic disorders.
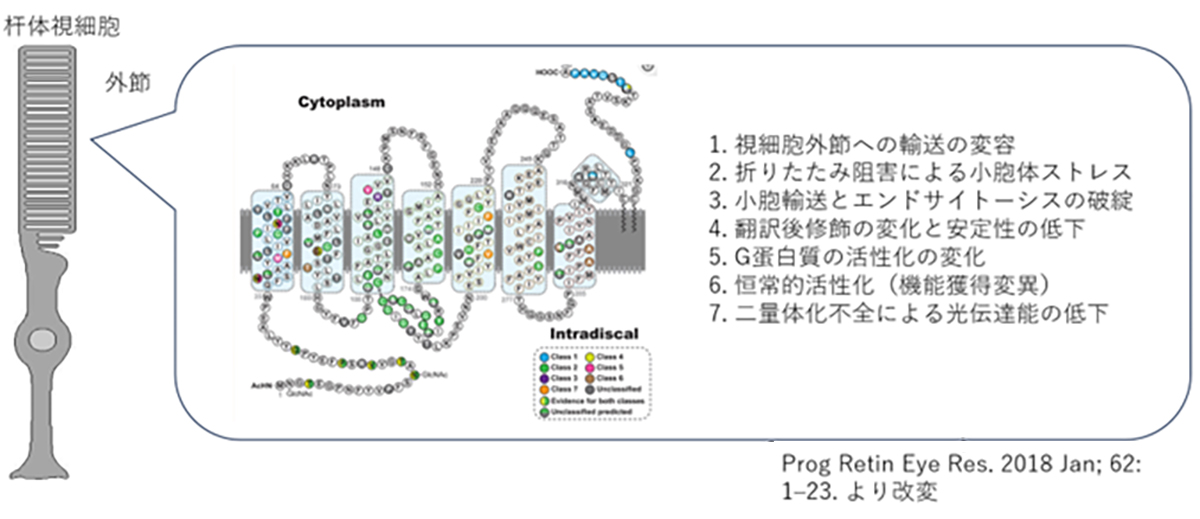
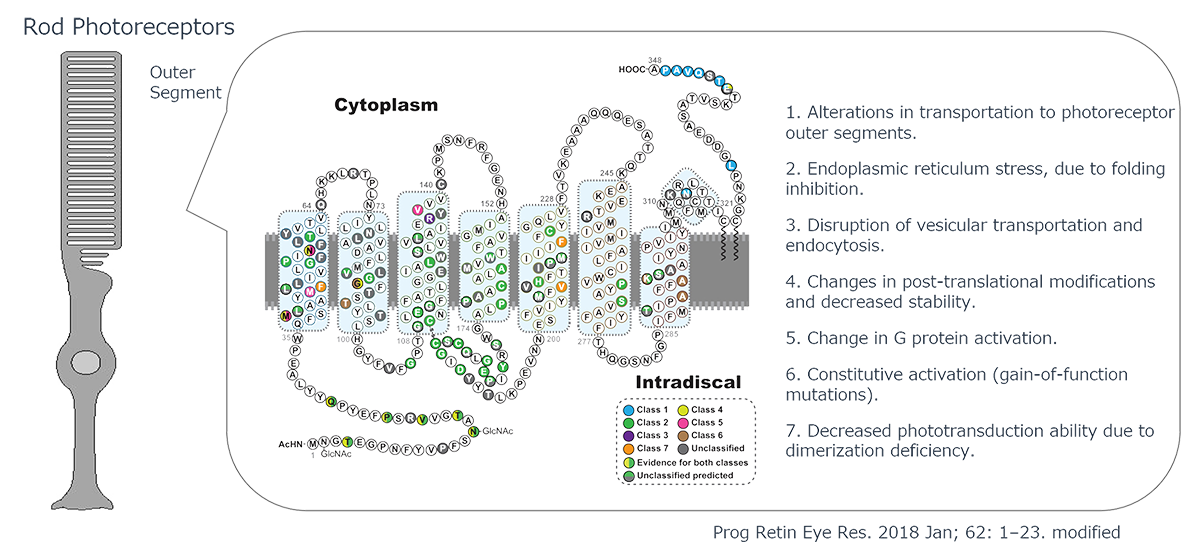
The Pathogenic Mechanisms of Rhodopsin Mutations in Photoreceptor Degeneration
Human Rhodopsin (RHO), a protein crucial for visual phototransduction, consists of 348 amino acids. Genetic analyses have identified more than 110 RHO-associated pathogenic mutations. Most of these mutations exhibit dominant-negative or gain-of-function characteristics, significantly altering protein function.
The functional consequences of these mutations are highly dependent on their location in the gene sequence. The observed effects include reduced photosensitivity, constitutive activation, and impaired intracellular trafficking of RHO protein. The severity of these inhibitory effects varies considerably among different mutations, necessitating precisely tailored therapeutic interventions through small-molecule compounds or other pharmacological agents.
Notably, RHO-associated pathologies can also arise from haploinsufficiency, in which a single functional allele is inadequate to maintain normal physiological function. The dual nature of RHO-related disorders presents a complex challenge for gene therapy approaches. Effective interventions must address both the correction of dominant-negative mutations and restoration of adequate wild-type RHO expression.
This multifaceted pathogenic mechanism underscores the need for comprehensive therapeutic strategies that can simultaneously mitigate the deleterious effects of mutant proteins and ensure sufficient levels of functional rhodopsins. This approach is critical for developing efficacious treatments for RHO-associated retinal degenerative disorders.
Genome editing-based therapy
Genome editing, a groundbreaking technique that allows us to rewrite the genetic code within cells, has tremendous potential for treating dominant-negative gene mutations. This innovative approach targets specific DNA sequences or mRNA transcripts in order to introduce precise genetic changes.
Our research is at the forefront of developing gene therapies with a strong emphasis on harnessing the power of genome editing. Through collaborative efforts with Hiroshima University, we have made significant strides in designing highly specific Zinc Finger Nucleases (ZFNs). These engineered ZFNs have shown remarkable efficiency in editing adult photoreceptor cells, rivaling the performance of the widely-used CRISPR-Cas9 system. We are also pushing the boundaries of gene therapy by developing treatments using Homology-Independent Targeted Integration (HITI) gene insertion techniques. This approach offers new possibilities for precise genetic modifications.
Genome Editing Methodologies
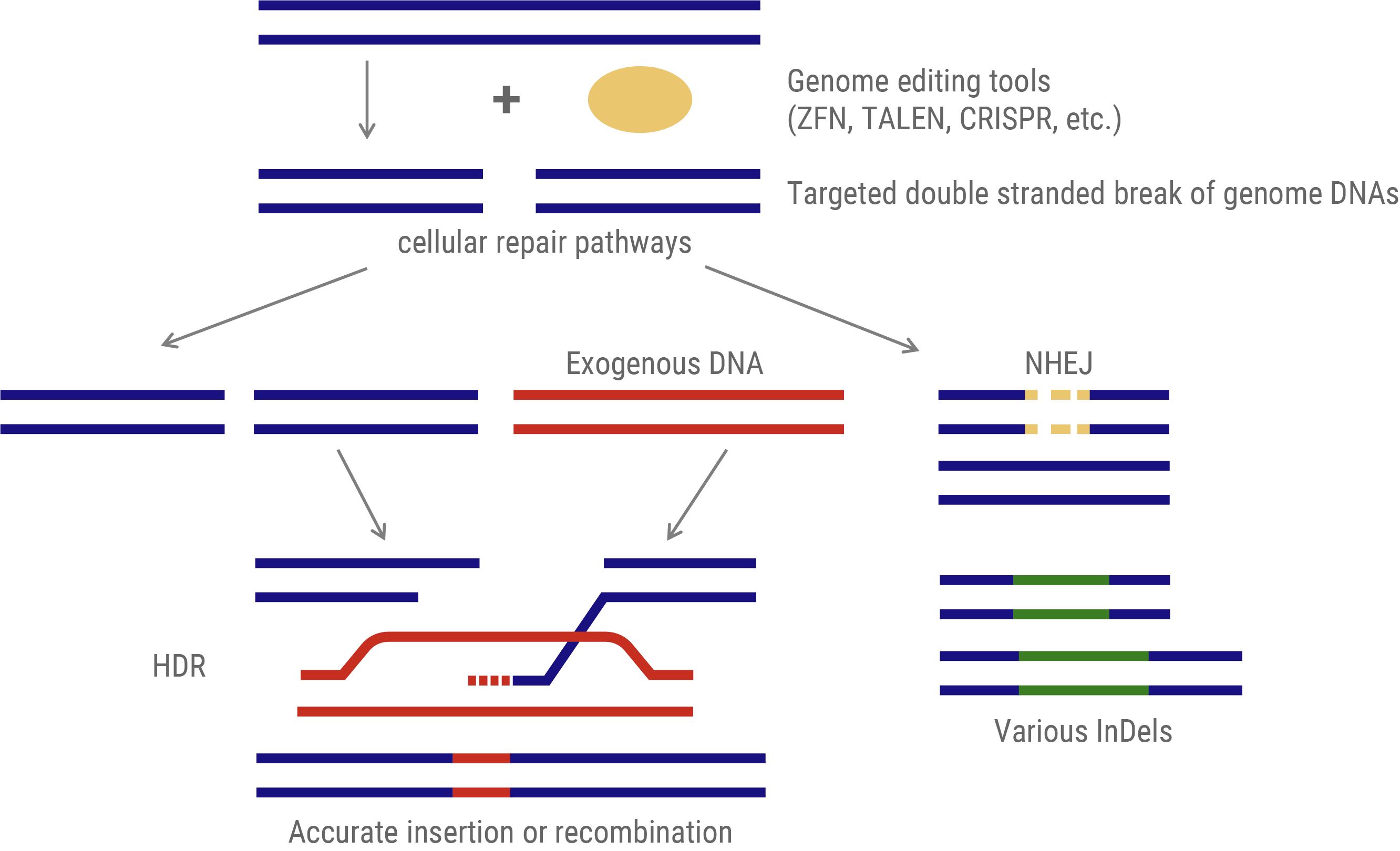
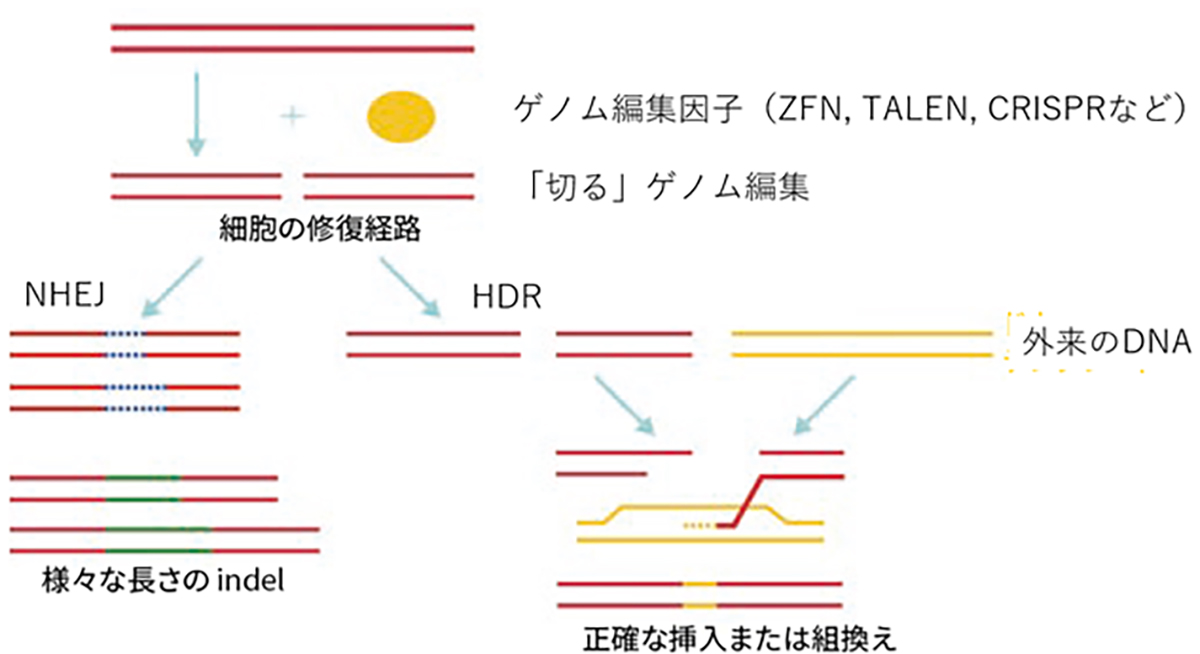
The predominant genome-editing strategy involves site-specific DNA cleavage at predetermined loci. These induced double-strand breaks (DSBs) activate two intrinsic cellular DNA repair pathways that can be exploited for gene insertion or targeted mutagenesis.
- Gene Insertion: This approach utilizes the homology-directed repair (HDR) mechanism. An exogenous DNA template, homologous to the target sequence but incorporating the desired modifications, is provided. During DSB repair, this template facilitates precise genomic alterations via recombination.
- Mutagenesis: This method leverages the non-homologous end-joining (NHEJ) pathway. NHEJ repairs DSBs by arbitrarily inserting or deleting nucleotides (InDels) at the break site. The stochastic nature of these InDels typically results in frameshift mutations or premature stop codons, which effectively disrupt the function of the target gene.
Genome editing via DNA repair machinery
Genome Editing Tools: Evolutionary Progression
Enzymes employed in genome editing have evolved through three primary generations.
- First Generation – Zinc Finger Nucleases (ZFNs): These heterodimeric nucleases utilize DNA-binding motifs derived from zinc finger transcription factors. Each motif recognizes a trinucleotide sequence, allowing for the modular assembly of target-specific nucleases.
- Second Generation – Transcription Activator-Like Effector Nucleases (TALENs): Derived from bacterial DNA-binding proteins, TALENs offer enhanced specificity. Each TALE motif recognizes a single nucleotide, enabling the design of highly specific genome-editing tools with extended recognition sequences.
- Third Generation – Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR): Based on prokaryotic adaptive immune systems, CRISPR technology comprises a Cas nuclease and single guide RNA (sgRNA). Unlike its predecessors, CRISPR employs RNA-DNA hybridization for target recognition, significantly simplifying target sequence design and accelerating genome editing research.
The advent of CRISPR has dramatically enhanced the accessibility and versatility of genome editing, catalyzing rapid advancements in the field of genetic engineering.
Highly efficient and Mutation-agnostic Gene Therapeutics to Treat Dominant Mutations
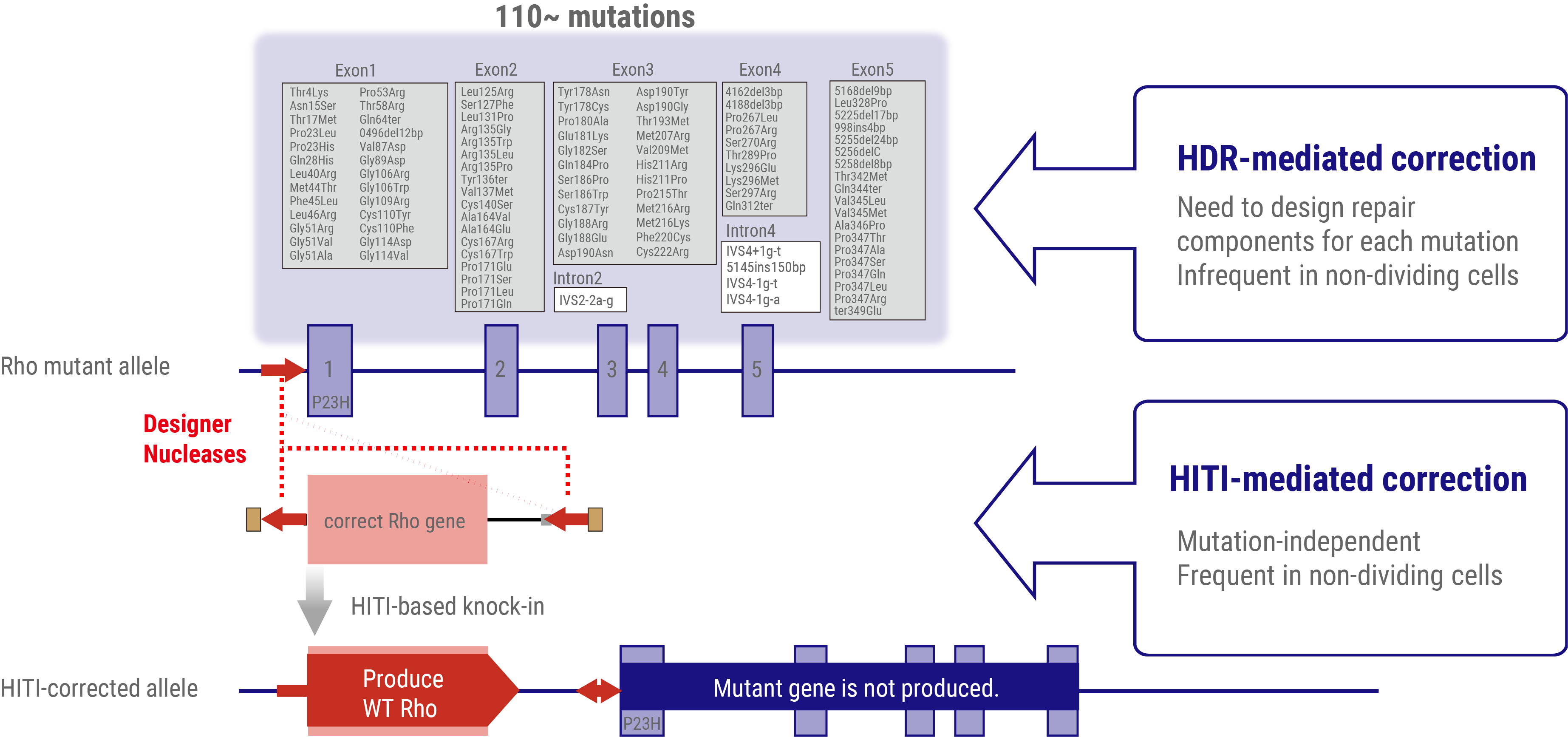
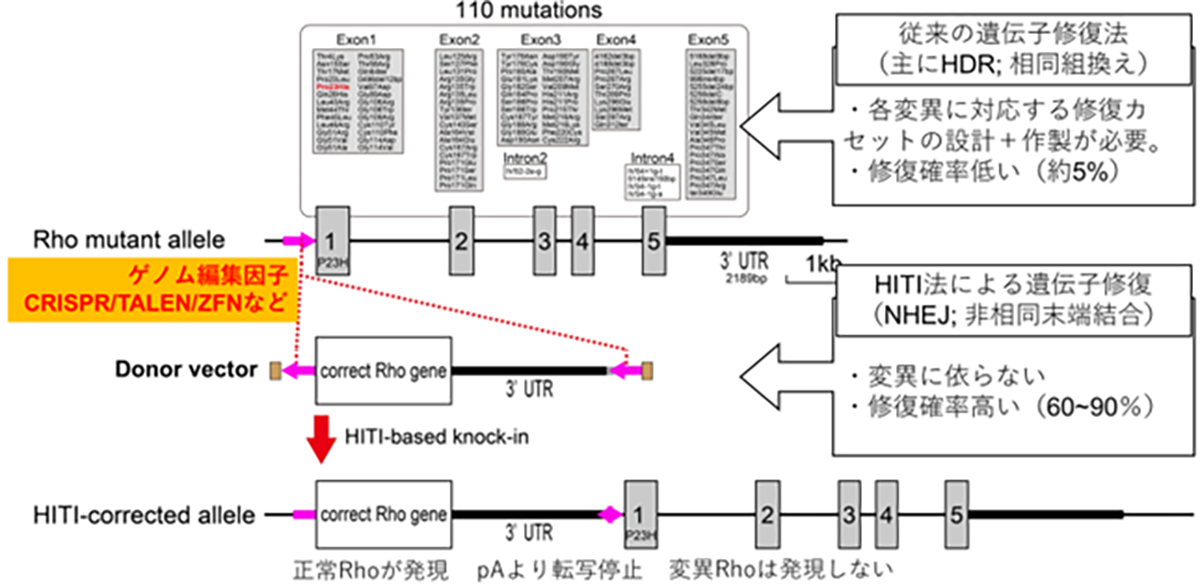
To address dominant mutations such as those affecting rhodopsin, a dual-action therapeutic approach is necessary. The gene therapy must simultaneously address the repair of the causative mutation inducing dominant inhibition and restore normal RHO gene expression. Given the presence of more than 110 potential mutation sites within the rhodopsin gene locus, a more efficient strategy involves the insertion or replacement of the full-length wild-type rhodopsin gene rather than developing mutation-specific therapies. This approach offers the potential to address all known mutations and accommodate newly discovered variants with a single therapeutic intervention.
Conventional approaches to introduce exogenous wild-type DNA sequences typically employ HDR-based genome editing. However, a significant challenge arises from the limited efficacy of HDR in post-mitotic cells, which constitute the majority of adult tissue cells, including the retinal neurons. To overcome this limitation, a homology-independent targeted integration (HITI) method has been developed. This innovative approach exploits the non-homologous end-joining (NHEJ) DNA repair pathway, which demonstrates high efficiency in non-dividing cells, to achieve highly efficient insertion of exogenous donor genes. Thus, HITI-mediated targeted gene therapeutics are ideal for targeting terminally differentiated adult cells such as retinal neurons.
By strategically positioning the wild-type sequence DNA immediately upstream of the start codon of the mutated gene locus, the expression of the normal gene is facilitated using the endogenous promoter, while concurrently suppressing the expression of the mutated allele. This elegant approach effectively addresses both the dominant-negative effects of the mutation and restores the wild-type RHO gene expression.
Our current research focuses on the development of HITI-based gene insertion therapeutics targeting the spectrum of rhodopsin mutations. This approach represents a significant advancement in the field of IRD therapeutics and offers the potential for a comprehensive treatment strategy for rhodopsin-associated retinal degeneration.
HITI-mediated gene-insertion therapeutics to treat RHO-associated patients
Zinc Finger Nuclease (ZFN)-mediated Genome Editing
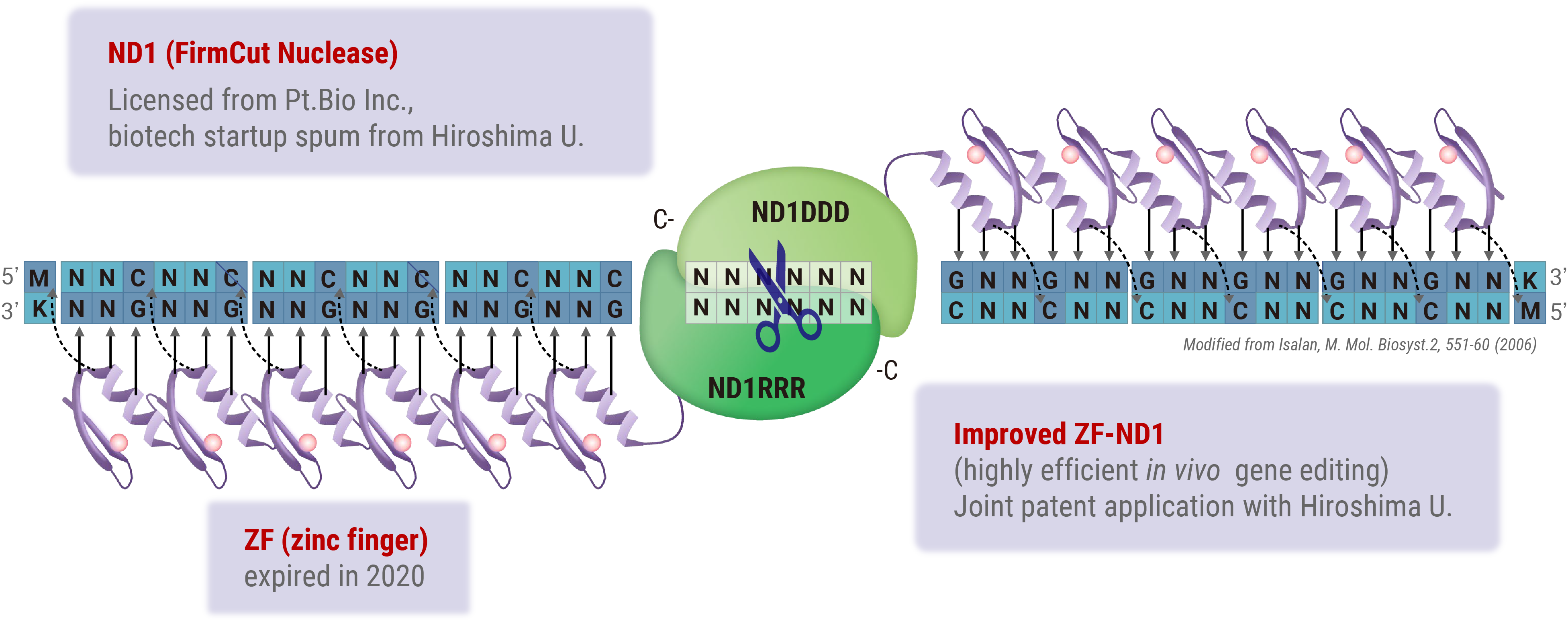
Zinc Finger Nucleases (ZFNs) offer a distinct advantage in gene therapy applications owing to their relatively low molecular weight, which facilitates efficient packaging into viral vectors, such as adeno-associated viruses (AAV). However, the design of highly specific ZFNs for target sequences has traditionally been complex and time-consuming. Through collaborative research with Hiroshima University, we have developed a high-throughput methodology for generating highly specific ZFNs. Utilizing Hiroshima University’s proprietary FirmCut nuclease ND1, we established a ZFN development technology that achieves cleavage efficiencies comparable to those of CRISPR-Cas9 systems in terminally differentiated retinal cells.
Several critical factors must be optimized in the development of therapeutic agents employing ZFN-driven HITI-mediated gene insertion. These include the design of constructs to maximize gene functionality, selection of appropriate vectors for gene delivery, and refinement of the insertion techniques. Our research leverages insights from functional genomics and stem cell biology to inform the design of constructs with optimized gene functionality.
In collaboration with Synplogen Co., Ltd., Kobe, Japan, we pioneered the development of high-quality AAV vectors. Furthermore, our partnership with Kobe Eye Center Hospital is focused on advancing our understanding and methodology of safe and accurate gene delivery operations in the context of retinal tissue.
Novel genome editing tool with ZF (zinc finger) and FirmCut nuclease
What is ZFN-Based Genome Editing?
Introduced in 1996, ZFN is the first-generation genome editing enzyme, an artificial protein blending the DNA-binding domain called the Zinc finger motif with the nuclease domain. A single Zinc finger can recognize three base pairs of DNA. By linking multiple Zinc fingers, you can produce a Zinc finger that recognizes extended DNA sequences and binds specifically to target sequences. As endonucleases operate as dimers, when two ZFNs bind to their respective target sequences, a nuclease dimer forms, resulting in DNA cleavage.
Improved ZFN
While ZFNs offer the advantage of a smaller molecular weight, making them easier to load onto vectors like AAV, designing a highly specific ZFN for a target sequence is challenging and time-consuming. However, our joint research with Hiroshima University has achieved high-throughput development of highly specific ZFNs. Leveraging Hiroshima University’s FirmCut nuclease ND1, we established ZFN development technology that achieves a cleavage efficiency comparable to CRISPR-Cas9 in adult retinal cells.
ZFN and Gene Delivery Techniques
In developing therapeutic agents through HITI gene insertion, it’s vital to craft the right constructs to ensure peak gene functionality, pinpoint the optimal vector for gene delivery, and hone the insertion techniques. Drawing from insights in functional genomics and stem cell development, our research diligently focuses on designing constructs for optimized gene functionality. Collaboratively working with Simprogen Corp. in Kobe, we’re pioneering the development of high-quality adenoviral vectors. Moreover, in partnership with the Kobe Eye Center Hospital, we’re advancing our understanding and methodology of precise gene insertion techniques.
Targeting All Mutations using the HITI Gene Insertion Method
For dominant mutations, such as those targeting rhodopsin, a gene therapy remedy needs to address both the repair of the causative mutation causing dominant inhibition and the replenishment of normal RHO gene expression. Since there are 110 potential mutation sites in the rhodopsin gene locus, it’s more efficient to insert (or replace with) the full-length normal rhodopsin gene rather than develop a separate gene therapy for each mutation. This way, one treatment can potentially address all mutations, including any new ones.
The HITI Method
When introducing external, normal-sequence DNA, the common approach utilizes HDR-based genome editing. A challenge arises since HDR is prevalent in dividing cells but less so in non-dividing cells, and the majority of adult tissue cells are non-dividing. To address this, the HITI (Homology-independent targeted integration) method was developed. It leverages the NHEJ repair pathway, notable for its high efficiency in non-dividing cells, to accurately insert foreign genes. This makes gene insertion in specific sequences of adult cells, such as retinal cells, achievable. By positioning the normal sequence DNA just prior to the start codon of a mutated gene locus, the normal gene is expressed using the original gene’s promoter, suppressing the mutated gene’s expression. Essentially, this method simultaneously rectifies dominant mutations causing dominant inhibition and restores the expression of the normal RHO gene.
Potential of Genome Editing for Dominant Mutations
Genome editing, which allows rewriting of cellular genetic information, holds great promise for treating gene mutations that manifest due to dominant inhibition. This technique targets specific sequences in DNA or its transcription product, mRNA, to introduce changes.
Genome Editing Tools
The enzymes employed in genome editing come in three main categories or generations:
- First generation – ZFN (Zinc finger nuclease): Based on various transcription factors’ DNA binding motifs, each motif recognizes three base pairs of DNA.
- Second generation – TALEN (Transcription activator-like effector nuclease): Originating from bacterial DNA binding motifs, each motif recognizes a single base of DNA. Compared to ZFN, TALEN recognizes longer sequences but allows designing high specificity genome editing tools.
- Third generation – CRISPR (Clustered regularly interspaced short palindromic repeats): Founded on the bacterial immune mechanism (CRISPR/Cas system), it consists of the Cas nuclease and a single-stranded guide RNA (sgRNA) recognizing the target sequence. Unlike ZFN and TALEN that use protein motifs for base recognition, CRISPR identifies target sequences through RNA-DNA interactions. This simplifies target sequence prediction and design, significantly advancing genome editing research.
Therapeutic Genome Editing Techniques
The commonly utilized genome editing approach involves “cutting” at the target sequence. The induced cuts exploit two intrinsic cellular gene repair mechanisms to insert (recombine) genes or introduce mutations.
- 1. Gene insertion: This approach uses the homology-directed repair (HDR) mechanism, where a repair gene is synthesized and recombined from an allele. During the cutting process, an externally provided template DNA with the correct sequence facilitates precise genome editing.
- 2. Mutation introduction: This method employs the non-homologous end-joining (NHEJ) mechanism. NHEJ repairs the cut DNA ends by arbitrarily inserting or deleting bases (InDels). As these InDels are random, they usually result in target gene modifications, primarily knockouts.
ZFNによるゲノム編集とは
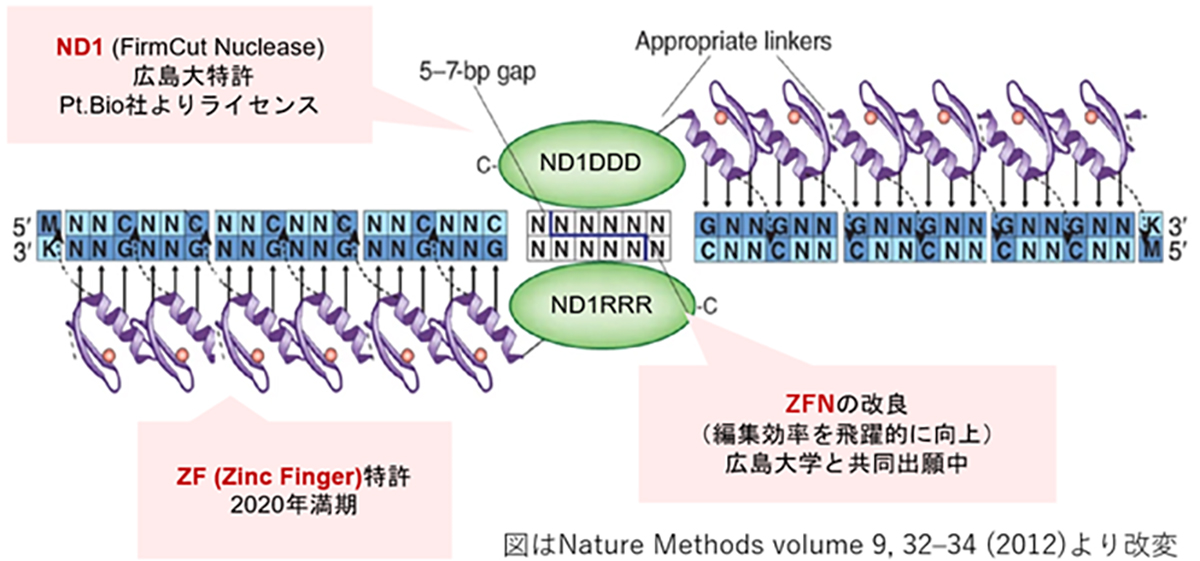
ZFNは1996年に発表された第1世代のゲノム編集因子で、Zinc fingerモチーフと呼ばれるDNA結合ドメインにヌクレアーゼドメインを結合した人工タンパク質です。1つのZinc fingerは3塩基対のDNAを認識するので、Zinc fingerを複数連結すると長いDNA配列を認識し標的配列に特異的に結合するZinc fingerを作製できます。ヌクレアーゼドメインは2量体で機能するエンドヌクレアーゼが用いるため、図のように2つのZFNを作製して各標的配列に結合した時にヌクレアーゼ2量体が形成される事によりDNAが切断されます。
改良ZFN(広島大学との共同技術開発)
ZFNは、CRISPRやTALENよりも分子量が小さくAAV等のベクター(運搬体)に載せやすい利点がある反面、目的配列に特異性の高いZFNの設計が難しく作製に時間を要するため、開発の大きな律速となっていました。我々は、広島大学との共同研究より特異性の高いZFN開発のハイスループット化を実現し、また同大学が開発したFirmCutヌクレアーゼND1を用いる事等により、成体視細胞でCRISPR-Cas9と同等の切断効率を持つZFNの開発技術を確立しました。
ZFN, 挿入遺伝子の送達技術(シンプロジェン社との協業・神戸アイセンターとの連携)
HITI遺伝子挿入による治療製剤の開発には、遺伝子が効率的に機能するためのコンストラクト設計、遺伝子を送達するベクター(運搬体)、そして導入手法の最適化が重要です。我々の研究では、機能ゲノム学と幹細胞発生学の知見を基にして、遺伝子が効率的に機能するためのコンストラクトを設計しています。また、シンプロジェン社(神戸市)と協力し、高品質のアデノ随伴ウイルスベクターの開発に取り組むとともに、神戸アイセンター病院と連携し適切な導入手法の確立のための研究を進めています。
図 ZFNの遺伝子認識イメージと、実験による編集効率の向上
HITI遺伝子挿入法による、全変異を対象とした遺伝子治療製剤開発
顕性型変異、例えばロドプシンを標的とした原因遺伝子治療製剤には、優性阻害となる原因変異の修復と、正常RHO遺伝子発現の補充の両方への対処が必要となります。ロドプシン遺伝子座には110カ所の変異があるため、各変異に対する遺伝子治療を個別に開発するよりも正常な全長のロドプシン遺伝子を挿入(置換)する方法がより効率的で、1種類の製剤で新規の変異を含む全ての変異を治療する事が可能となります。
HITI法:非分裂細胞で高効率な遺伝子挿入を実現する技術
正常配列DNAを外から供給する場合はHDRを介したゲノム編集技術が一般的ですが、HDRは分裂細胞での頻度が高く非分裂細胞では低い傾向があります。成体の組織を構成するほとんどの細胞は非分裂細胞であることから、成体細胞に対してのDNAを導入・置換の効率が低い事が課題でした。HITI(Homology-independent targeted integration)法による遺伝子挿入法は、非分裂細胞でも頻度が高いNHEJ修復経路を利用して外来の遺伝子を正しい方向で挿入する技術であり、網膜細胞を含む成体細胞でも高頻度で目的配列に遺伝子を挿入する事が可能となります。変異を持つ遺伝子座の翻訳開始部分(開始コドン)前に正常配列DNA挿入する事で、遺伝子本来のプロモータから挿入した正常遺伝子を発現させつつ、変異遺伝子の発現が停止します。すなわち、顕性型変異における優性阻害となる原因遺伝子の修復と正常RHO遺伝子発現の補充の両方の対処が可能となります。
HITI遺伝子導入の図解
顕性型変異に対するゲノム編集の有用性
顕性遺伝型のうち特に優性阻害により発症する遺伝子変異に対しては、ゲノム編集技術による治療法が高い効果を期待されています。ゲノム編集は細胞の遺伝情報(ゲノム)を書き換える技術の総称であり、ゲノム本体であるDNAや転写産物のmRNAの特定の配列を狙って変化させることが可能となります。
ゲノム編集因子
ゲノム編集を行う酵素はゲノム編集因子と呼ばれ、標的配列をデザインできるゲノム編集因子は第1世代のZFN(Zinc finger nuclease)、第2世代のTALEN(Transcription activator-like effector nuclease)、第3世代のCRISPR(Clustered regularly interspaced short palindromic repeats)に大別されます。ZFNは様々な転写因子のDNA結合モチーフが基となっており、1つのモチーフで3塩基対のDNAを認識します。TALENは細菌由来のDNA結合モチーフが基となっており、1つのモチーフで1つのDNAを認識するため、ZFNより配列が長くなりますが特異性の高いゲノム編集因子の設計が可能となりました。CRISPRは細菌の免疫機構(CRISPR/Casシステム)が基になっており、Casヌクレアーゼと標的配列を認識するsgRNA(1本鎖ガイドRNA)で構成されます。タンパク質モチーフで塩基配列を認識するZFN, TALENに比べ、CRISPRは標的配列の認識をRNA-DNA相互作用によって行うため、標的配列の予測や構築が容易になりゲノム編集の研究応用が飛躍的に進みました。
ゲノム編集による治療方法(変異の修復方法)
現在頻用されるゲノム編集は狙った配列を「切る」編集で、切断に伴い誘導される細胞内の2つの遺伝子修復機構を利用して、遺伝子を挿入(組み換える)編集や変異を加える編集を行います。遺伝子を挿入する編集は、相同組換え修復(homology-directed repair; HDR)と呼ばれる対立遺伝子から修復遺伝子を合成し組換える修復を利用し、切断時に正常配列を持つ鋳型DNAを外来から供給し組み替える事で正確なゲノム編集が期待できます。変異を加える編集は、非相同末端結合(non-homologous end-joining; NHEJ)と呼ばれる、切断されたDNAの両端を任意の塩基の挿入・欠失(InDel)を伴いながら結合する修復機構を用います。InDelは任意であるため標的遺伝子の改変(主にノックアウト)が起こります。
図:ゲノム編集因子と、「切る」ゲノム編集からできる事(HDRとNHEJ)
網膜におけるロドプシンの役割
ロドプシン(RHO)は杆体視細胞の外節と呼ばれる部分に局在する光受容タンパク質で、僅かな光でも感知できるように遺伝子が進化しており、また外節部分の総タンパク質の9割をロドプシンが占めています。杆体視細胞が夜間や暗いところで光を感知する特徴はロドプシンの機能に大きく依存しています。ロドプシン(RHO)遺伝子変異は顕性型の網膜色素変性の原因遺伝子の約3割を占め、世界では10万人程度の患者さんが推定されています。
変異ロドプシンによる網膜色素変性の発症機構
ヒトのロドプシンは348のアミノ酸から構成されていますが、これまでの遺伝子診断より110カ所の異なる変異が報告されています。変異遺伝子のほとんどが優性阻害/機能獲得変異を示しますが、機能変容は変異場所による事が知られており、光感受性の低下や恒常的活性化やロドプシンタンパク質の輸送不全などがあります。また阻害効果も変異により異なるため、低分子化合物などを用いての対症療法には適切な薬剤処方が必要です。加えてロドプシン遺伝子はハプロ不全でも発症する原因遺伝子であるため、RHOを標的とした原因遺伝子治療製剤には優性阻害となる原因変異の修復と、正常RHO遺伝子発現の補充の両方への対処が必要です。